Differential Peak Calling of ChIP-Seq Signals with Replicates with THOR
Manuel Allhoff, Kristin Seré,Juliana Pires, Martin Zenke and Ivan G. Costa
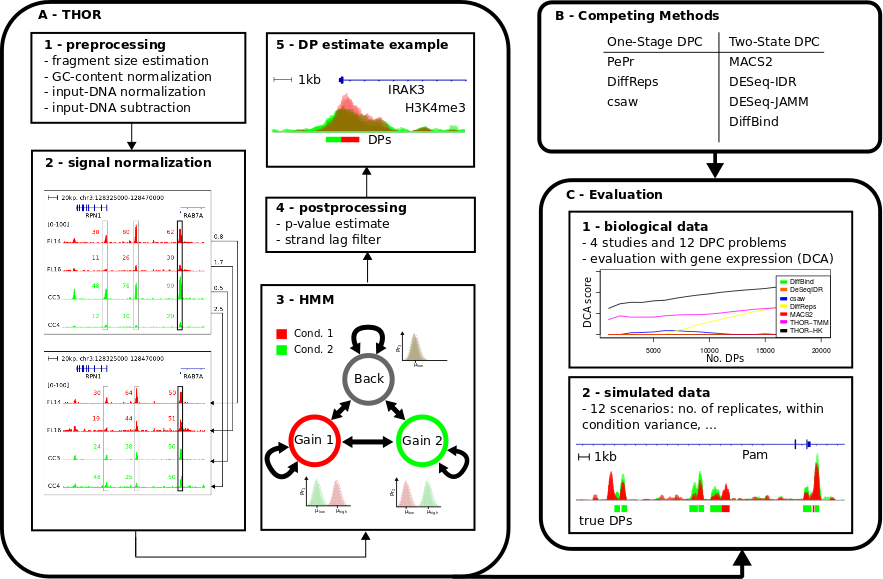
Method
The study of changes in protein-DNA interactions of dynamic systems, such as cell differentiation, response to treatments or the comparison of healthy and diseased individuals, is still an open challenge. Of particular relevance is the analysis of ChIP-seq data in clinical scenarios, as they allow detection of epigenetic markers and regulatory SNPs. Detecting regions with changes in ChIP-seq signals between two distinct biological conditions is called differential peak calling. However, there are few computational methods performing differential peak calling when conditions have replicates. Moreover, none of these previous approaches addresses ChIP-seq specific experimental artefacts arising from studies with biological replicates.
We propose THOR, a Hidden Markov Model based approach, to detect differential peaks between pairs of biological conditions with replicates. THOR provides all pre- and post-processing steps required in ChIP-seq analyses. Moreover, we propose a novel normalization approach based on house keeping genes to deal with cases where replicates have distinct signal-to-noise ratios. To evaluate differential peak calling methods, we delineate a methodology comprising the use of both biological and simulated data. We evaluate THOR and seven competing methods on data sets with distinct characteristics from in vitro studies with technical replicates to clinical studies of cancer patients. Our evaluation analysis of 12 differential peak calling problems indicates that THOR performs well in all scenarios, in particular in cases where ChIP-seq replicates have larger signal variance.