Detection of Epigenetic Traits from Next Generation Sequencing Data
 We develop methods for detection of epigenetic features of cell, such as cell active binding sites from open chromatin essays and chromatin changes during biological processes. These methods include the integrative analysis of NGS data as DNase-, ChIP-, and RNA-Seq and single cell variants of these protocols. In this context, we develop a toolbox to perform several basic computation tasks required for the analysis of regulatory genomics data. The toolbox also includes motif matching from DNA sequences, signal processing of next generation sequencing, manipulation and graphical analysis of genomic intervals and profiles and is available at www.regulatory-genomics.org .
We develop methods for detection of epigenetic features of cell, such as cell active binding sites from open chromatin essays and chromatin changes during biological processes. These methods include the integrative analysis of NGS data as DNase-, ChIP-, and RNA-Seq and single cell variants of these protocols. In this context, we develop a toolbox to perform several basic computation tasks required for the analysis of regulatory genomics data. The toolbox also includes motif matching from DNA sequences, signal processing of next generation sequencing, manipulation and graphical analysis of genomic intervals and profiles and is available at www.regulatory-genomics.org .
People
Eduardo Gade Gusmao (former member)
Manuel Allhoff (former member)
Selected Publications
Gusmao E.G., Allhoff, M., Zenke, M., Costa, I.G. (2016), Analysis of computational footprinting methods for DNase sequencing experiments,Nature Methods, 13, 303–309 [paper][pre-print][supp].
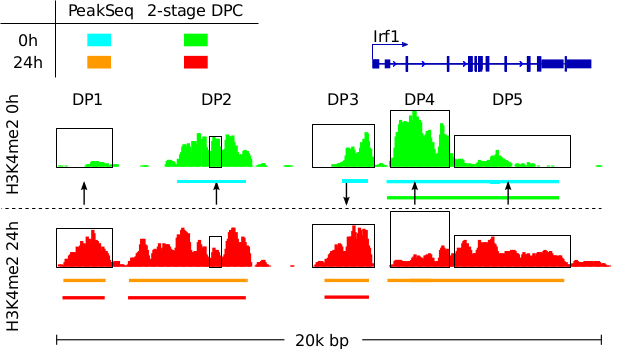
Allhoff, M., Sere K., Freitas, J., Zenke, M., Costa, I.G. (2016), Differential Peak Calling of ChIP-seq Signals with Replicates with THOR, Nucleic Acids Research, epub gkw680 [paper][supp][data].
Allhoff, M., Sere, K., Chauvistre, H., Lin, Q., Zenke M, Costa IG, Detecting differential peaks in ChIP-seq signals with ODIN, Bioinformatics, 30, 3467-3475 [paper][data][supp].
Gusmao EG, Dieterich C, Zenke M, Costa IG, Detection of Active Transcription Factor Binding Sites with the Combination of DNase Hypersensitivity and Histone Modifications, Bioinformatics, 30(22):3143-51. [paper] [Supp].